생명공학 관련 일을 하시는 분들이라면 다들 pcr과 western blot에 대해서 들어보셨을 것이다. 앞서 pcr법이 아주 좋은 조회수를 기록해주었다. 이제 종강할 때가 되니 조회수가 줄어들긴 했다. 대학생 여러분들... 공부는 학기중에만 하면 끝날줄 알았죠? 이모는 삭신이 쑤시는 나이가 되었는데도 공부를 한답니다... 화이팅!
2022.03.01 - [기초/생명과학 생명공학 Biotechnology] - PCR의 정의와 기본 원리, PCR, rt-PCR, real time PCR, qPCR 차이점
거두절미하고, 웨스턴블랏, 웨스턴블롯, 오늘은 이러한 블랏 방법을 왜 사용하는지, 무엇이 중요한지 말하려고 한다.
태초에 남쪽에서 블랏이 일어났다. 서던 블랏

Sanger sequencing이 있기전 서던이라는 사람이 dna염기서열을 알아내기 위해서 개발한 방법이 서던 블랏이다. DNA를 절단해서 그걸 크로마토그래피법을 이용해서 전기영동을 내린 뒤에, 특정 membrane에 붙인 다음 방사선 동위원소 등(옛날엔... 요즘은 bionylated된게 다 나온다)으로 보고 내가 원하는 DNA샘플에 이 염기서열이 있는가?! 를 보는 것이다. 이때 gel에서 membrane으로 옮기는 과정을 거치는 것을 blot이라고 한다. gel 에 있던 정보를 membrane으로 띄우는 것, 그것이 blotting이다.

이 분이 Edwin Southern이시다. 아직 살아계심... 분자생물학이 얼마나 최신 기술인지 알수 있는 부분이다. 이 분께서 DNA를 이용한 southern blot을 만드신 후에 상보적인 RNA에 대한 Northern blot이 만들어졌고 (북쪽) 그리고 서쪽인 Western엔 단백질을 분석하는 Western blot이 생기게 되었다. ㅋㅋㅋ... 이런 박사식 유우머로 인해 단백질을 분리해내는 웨스턴 블랏이 생기게 되었음. 그리고 최근에는 단백질 변형을 살피는 서쪽의 반대쪽.. 동쪽에 eastern blot이 생겼다. HAHAHA..
웨스턴 블롯, 왜하는 걸까?
우리는 앞서 PCR법에 대해서 살펴봤다. 내가 원하는 DNA가 얼마나 있는지 보기 위해서 qPCR을 사용했다. PCR로 정량까지 가능해졌는데, 왜?! 왜!!! 굳이!! 거지같은 블롯팅을 하는 걸까?
그건 생체내에서 모든 것은 단백질로 연결되기 때문이다.

우리가 살펴봤던 DNA, RT-PCR을 해서 봤던 RNA, 결국에 최종 산물은 단백질이기 때문이다. RT-PCR을 해서 체내에 mRNA가 얼마나 있는지 qPCR로 정량을 해봤다고 해도, 결국에 체내에 단백질 산물이 돌아야 생화학적인 반응이 있다고 봐도 무방하다. 그래서 우리는 단백질이 진짜 많이 생겼냐? 덜생겼냐? 를 보기 위해서 웨스턴 블롯팅을 한다.
웨스턴 블롯을 하면 알게 된다. qPCR은 손을 덜 타는 실험이고... 돈이 덜 드는 실험이었음을..
western blot의 전반적인 과정

1. 단백질에 음전하 끼얹기 Running
단백질은 다양한 아미노산으로 이루어져 있다. 이때 산성 아미노산이 많으면 음전하를 띄고, 염기성 아미노산이 많으면 양전하를 띈다. gel electrophoresis를 하기 위해서 전류는 -에서 +로 흐르기에, 모두 음전하를 주는 방식이 필요하다. 그걸 해주는 친구가 바로 계면활성제로 내리는 SDS PAGE다.
SDS page를 이용해서 단백질을 전기영동 하여 크기별로 분리한다.
2. membrane에 옮기기 Transfer
이제 내가 원하는 단백질이 얼마나 있는지 보기 위해서 membrane에 옮긴다.
3. blocking, primary antibody, secondary antibody
내가 누누하게 말했던 것 처럼 항체를 이용한 실험은 거의 indirect이다. western blot도 마찬가지로 indirect가 상용화 되어있다. 5% skim milk를 이용해서 비특이 반응을 줄인다음, 1차 항체를 붙이고 2차항체를 붙인다.
2023.01.06 - [응용/면역학 Immunology] - 면역염색 원리와 종류, 프로토콜 (IHC, Immunohistochemistry)
면역염색 원리와 종류, 프로토콜 (IHC, Immunohistochemistry)
면역염색의 역사 실험을 하다 보면 많이 하게 되는 염색 중 하나인 IHC, 면역 염색은 내가 원하는 특정 단백질이 고정된 조직 안에서 존재하는지를 항체를 붙여 확인하는 방법이다. 현재까지도
labanimal.tistory.com
4. 현상 ECL
2차항체에는 HRP가 붙어있다. 그래서 Luminol과 과산화수소를 반응시켜 빛이 나오고, 이를 사진을 찍어내는 방식으로 관찰할수 있다.

옛날에는 xray필름으로 봤지만, 요즘은 좋은 chemidoc이 많이 나와서 이를 이용해서 사진을 찍을 수 있다.
Western blot protocol, gel recipe
western blot protocol이나 gel recipe같은 경우 대대로 실험실마다 내려오는 맛집 비법처럼 ... 비법이 있기 때문에 환경에 따라 trouble shooting을 해가면서 하시면 됩니다.
1. 단백질 뽑기
sample에서 protein을 추출해야한다. 이때 쓰는 lysis buffer는 다양한 종류가 있다. 계면활성제의 구조상 특징 때문에 생기는 다양함이라고 보면 된다. 단백질을 최대한 보존해야 하는 immunoprecipitation(IP)과정에는 native buffer를 쓰고, western blot을 할 때는 denaturing buffer를 쓴다.
western blot에서 보통 조직/세포에서 뽑을 때 쓰는 RIPA buffer는 Triton-X100이라는 계면활성제와 SDS가 들어가있다.
CST RIPA buffer
RIPA Buffer (10X) | Cell Signaling Technology
For Research Use Only. Not for Use in Diagnostic Procedures. © 2024 Cell Signaling Technology, Inc. All rights reserved.
www.cellsignal.com
우리는 이 제품을 사용중이고, EP tube에 1ml씩 소분하여 냉동보관 했다가 필요할 때 10배 희석해서 냉장보관 짧게 해서 사용중이다. 근데 이렇게 사용한 RIPA 버퍼도 gel에 내릴때 좀 가벼운 면이 없지않아 있어서 요즘은
intron proprep
인트론바이오 DR파트
Protein PRO-PREP™ Protein Extraction Solution (C/T) PRODUCT INFORMATION Description 세포나 조직으로부터 빠르고 간편하게 고순도의 total protein을 추출 할 수 있는 제품 • 세포나 조직으로부터 추가 조작 없이 단
www.intronbio.co.kr:6002
을 사용중이다. 이쪽이 밀도가 높아서 피펫팅할때 양 차이가 날 일이 적고, 특히 지방이 많이 낀 샘플에서 좀 더 깨끗하게 단백질을 뽑을 수 있어서 선호하는데 비싸긴 하다. RIPA 와 병행해서 사용할 수는 없음. 정량에서 차이가 남.
- 조직/세포에 적정량의 buffer 넣고 homogenize, 나는 좀 옅게 많이 뽑아서 ul당 왔다갔다 하는 양이 적도록 하는 것을 선호한다. 곰손이라.
- ice에서 30분 정도 cool incubation
- 원심분리 14,000rpm 20min, 원심분리기 마다 각도와 거리 다르므로 실험실 조건 확인
- 지방층 (상층) 과 debris 제외한 부분을 딴다. (500ul 정도의 buffer 넣었으면 150ul정도만 딸수 있도록)
- 나는 곰손이라 300ul 정도를 따고 한번 더 원심 돌려서 150ul을 딴다.
2. 단백질정량
이제 단백질을 정량해야 한다. 단백질 정량법도 종류가 다양하지만, 실험실에서 많이 쓰시는 방법으로는 bradford법과 BCA법이 있겠다. 어떤 방법이든 간에 BSA 표준 곡선과 내 흡광도를 비교해서 단백질을 정량하는 방법임은 다른게 없지만, 반응하는 시약이나 흡광하는 파장, 그리고 시험자의 선호도에 따라 나뉜다고 보면 된다.
실험실에 있는 분광광도계의 detection 가능한 파장을 확인 한 뒤 보통 세팅을 해놓기 때문에 ㅋㅋ 실험실에 세팅 되어 있는 방법으로 하면 된다. 우리 실험실은 Bradford법을 사용중이다. 근데 사실 Bradford 시약인 Coomassie Blue가 온만데 묻어도 다 파란색으로 변함.... 바닥에 묻어도 파랗게 되고, 피펫팁에 묻어도 파란색이 된다. 이게 맞나? 싶어서 그래서 못믿겠다며 BCA법을 사용중인 곳도 많다.
- 96well plate에 dye 200ul, sample 5ul 넣기_우리는 따로 top down 안시킨다. 원심 넣을때 넘치는 불상사가 너무 많이 일어난다는 이유. ㅠㅠ loading할때 샘플 진짜 들어가는지 잘 보고 할 것.
- 최소 10분 room tempt. incubation, 여름엔 10분, 겨울엔 15분 해줌.
- 595nm에서 흡광도 측정, 분광광도계 shaking 10초, settle down 15초 해서 찍기
- BSA 표준곡선과 비교하기
- 정량해서 희석할 것 계산하기.
단백질 정량 계산 법

만약에 bsa 표준 함수가 이렇게 나왔다. Standard는 사실 온도나 습도에 따라 다르게 나올수 있어서 할 때마다 새로 하는 걸 추천은 하나, 많이 하는 곳에서는 여름/겨울에 한번씩 새로 하거나 실험하는 실험자별로 standard curve수식을 가지고 있는 경우가 많다.
만약에 내 샘플의 od값이 0.5로 나왔다고 치자.
0.5=0.06x-0.015
0.06x=0.615
x=10.25
그렇다면 나의 단백질 농도는 10.25ug/ul이다
이제 내가 쓸 well에 30ug의 protein을 30ul로 만들어 분주하고 싶다. 이 때 loading buffer는 4x짜리를 쓴다. 양을 30ul만 만들면 필히 모자라기 때문에 나는 36ul를 만들어 넣을 것이다. loading buffer가 4x이므로 나는 9ul의 loading buffer를 넣고, 27ul인데, 30ug이 들어가있는 단백질 샘플을 만들어야 한다.
우선 샘플의 농도가 10.25ug/ul이므로

sample 2.93ul
lysis buffer 27-2.93=24.07ul
loading buffer x4=9ul
총합 36ul 로 맞춘다.
sample buffer, loading buffer는 샘플이 얼마나 내려갔는지 보여주는 지표 역할을 하고 glycerol이 들어있어 well에 넘치지 않도록 해주며, SDS를 넣어 단백질이 음전하가 생기도록 한다. 보통 유통되는 sample buffer에 DTT가 들어있어 단백질의 이황화 결합을 깨주어 작게 만들어주지만, 우리는 900ul의 sample buffer에 β-Mercaptoethanol을 추가하여 95도씨에서 5분 가열하여 단백질을 환원시킨다.
- 샘플 희석 후 loading buffer넣기, 점성 있으므로 피펫팅 유의
- 95도씨 5분 가열
- 가열후 얼음에 cooling 곧장 하고 top down 시키기
3. Gel 만들기 및 running
단백질은 그 크기가 작기 때문에 보통 PCR후 전기영동 내렸던 agar를 사용하지 않는다. agar는 그 크기또한 불안정한 편이라 (요즘 좀 괜찮다고 하는데도) 단백질 내리기엔 적당하지 않아서 쓰는 것이 acrylamide이다. 얘네는 농도를 조절해서 pore size를 조절할수 있는 장점이 있다.
acrylamide를 얼마나 진하게 넣을 거냐, 전기 영동 할 때 buffer를 어느걸 쓸거냐에 따라서 실험실의 gel 조성과 buffer조성이 달라진다.
보통의 SDS PAGE는 Tris/Glycine/SDS buffer를 사용한다. 여기에서 Glycine은 pH에 따라 전기적으로 중성이냐 음성이냐가 결정된다. 우리가 보는 gel의 윗쪽은 pH가 중성, 아랫쪽은 ph가 염기성이다. 이때의 glycine은 윗쪽에선 중성, 아랫쪽에선 음성을 띄게 된다. 윗쪽에서 중성을 띄게 된 gylcine은 전류가 흐르는데 전기적으로 중성이다 보니, 갈곳이 없어서 가만히 있는다. 음전하를 띄는 우리의 샘플은 내려오는데, glycine때문에 쌓여서 모이게 된다. 이것이 stacking gel의 필요성이다.

이제 염기성인 resolving gel, running gel로 내려오면 glycine은 음전하를 띈다. 다른 단백질보다 빨리 빠지기 때문에 glycine이 스타트를 끊어주면 단백질이 내려오고, 큰 단백질은 천천히 내려와서 위쪽에, 작은 단백질들은 빨리 내려와서 아래쪽에 위치한다. 그러면 크기별 단백질 분리가 끝난다.
SDS PAGE | Stacking vs Resolving gel - YouTube
Stacking gel recipe, 5ml 기준
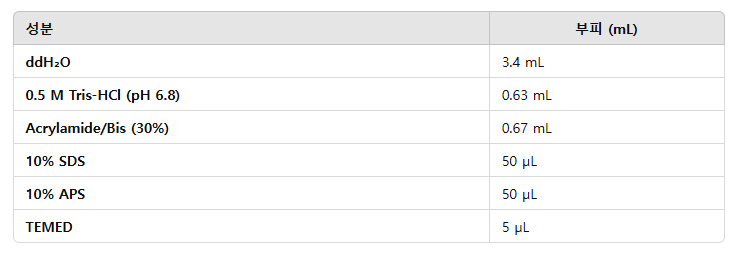
APS와 TEMED는 gel을 굳게 하는 역할이다. 제일 마지막에 넣기
Running gel recipe, 10ml 기준

보통은 10~12%를 많이 사용한다. 분자량에 따라 다르지만 대체로 12%인 듯. running gel 넣고 isopropanol 넣어 평평하게 만든 뒤 10분정도 굳히고 stacking gel 쌓은 뒤 comb를 꽂아 사용.
우리는 Glycine buffer를 사용하지 않고 MOPS를 사용하는 Bis-Tris buffer system을 사용중이다. MOPS는 pH변동 없이 하나의 pH로만 내릴수 있어서 더 안정적이고 깔끔하다고는 하신다. 대신 좀 비싸다. 우리는 심지어 premade를 구매해서 쓰는데 gel 하나 내릴때 마다 손이 달달 떨릴 정도. 뭐... APS나 TEMED를 까먹는다거나 여러 실수들이 잦아서 precast gel을 쓰게 된거겠지만, 고도로 숙련된 분들은 유리에 직접 gel을 만드는 것을 선호하신다. 신선한 것이 좋다며...
샘플을 loading후 보통 80~100V에서 1시간 30분 가량 running한다. 이때 저항이 발생하면 암페어 값이 높아지므로 midi gel 기준 2장 running 할 때 0.1A를 넘지 않는지 체크. 0.06A정도가 보통.
4. Transfer
이제 gel에서 내가 이용이 편한 membrane으로 단백질을 옮겨 주어야 한다. 이것이 Transfer 과정이다. 이때는 membrane에 단백질이 찰싹 달라붙어야 해서, 미끌거리는 SDS와 각종 계면활성제가 없는것이 좋다. 그래서 넣는것이 methanol.
transfer buffer는 보통 TPBS에 methanol을 넣어 준비한다. methanol이 휘발성이기에 날아가는데?? 다들 Transfer buffer는 색깔이 맛탱이가 갈 때까지 재사용한다.
이제 내가 단백질을 붙일 membrane도 종류가 여러가지이다. 보통은 NC를 많이 사용하고, 작은 단백질 붙이기, 깔끔한 밴드가 좋다! 한다면 PVDF를 사용하는 경우도 많다. 당근 PVDF가 비싸다. 멤브레인은 gel 크기에 맞게 잘려져 있는 것들도 있고, 롤로 판매되는 것도 있다. 크기에 맞게 잘려져 있는 membrane의 경우 filter paper가 함께 내장되어 있엇 습기, 건조에 조금 더 강하다. 대신 비쌈. ^^... 내가 잘라쓰는게 싸긴 싸다. 그래서 웨스턴을 많이 안하는 곳> 잘려진 membrane, 웨스턴 공장> roll membrane 사용을 한다.
여기에서 눈치챘겠지만 TPBS에는 SDS가 안들어 가는게 보통이다. 전하를 띄게 도와주는 물질이 부족하다보니, 저항값이 높아 열이 발생한다. Transfer buffer는 그래서 냉장보관 하고, 할 때도 ice pack을 넣어서 온도가 올라가는 걸 막아줘야한다. 잘못해서 buffer가 보글보글 끓으면 gel이 용해되어 단백질이 다 사라지는 기깔나는 경험 가능.
전류는 음에서 양으로 흐르므로
카세트 검은색>스펀지>필터>젤>멤브레인>필터>스펀지>빨간색
으로 쌓아야 한다. 그래야 젤에서(음)>멤브레인(양)으로 이동한다. 이 때 롤러로 살살 하지만 확실하게 문질문질 해줘야 공기방울이 안생긴다.
우리는 Transfer를 40~60V로 1시간 15분~30분 정도 옮긴다. 단백질 크기, 실험실 세팅, 버퍼 종류마다 다르므로 확인해보고 세팅값으로 진행하면 된다. 이 때 저항이 높지만 0.3A이상 올라가지 않도록 주의. 0.5를 넘으면 반신욕 ssap 가능할 정도임.
Transfer가 끝나면 membrane을 염색해서 진짜 단백질이 잘 붙었는지 봐야하는데 염색약으로는 보통은 Ponceau S를 사용한다. 빨간색은 Pinceau S고 파란색은 보통 CBB염색이다. 둘다 염색하고 나서 염색약을 다 빼고 항체를 붙여햐 한다는 불편함이 있으며, Ponceau S는 단백질 양이 적으면 안보이는... 단점이 있다. 그리고 뭔가 계속 헹구고 헹구고 헹궈도 불그스름한게 남아 있는 것 같아서 확실하게 염색이 다 빠졌네! 가 보이는 걸 좋아하는 실험실에선 CBB를 사용하기도 한다,

참고로 핵내 단백질을 검출 할 땐 보통의 Housekeeping 단백질을 쓸 수 없으니 Ponceau S 염색 사진을 기준으로 한다.
- Running 100V (0.1A이하) 1시간 30분
- Transfer 검>스펀지>필터>젤>멤브레인>필터>스펀지>빨 60V 1시간 30분, 0.3A이하, 아이스팩 필요시 중간 교체
- 필터와 젤 버리고 멤브레인 염색 (Ponceau S)
- 실험을 빨리 하고 싶으니 크기별로 잘라서 쓰기도 하는데, western이 망하는 제일 큰 이유는 가위질이다. ㅋㅋㅋㅋㅋㅋㅋㅋㅋㅋㅋㅋ 넉넉하게 가위질 해서 큰쪽/작은쪽 분리해서 사용해볼 것,
- PBS+0.1%Tween20으로 탈회
5. Blocking과 Primary antibody 붙이기
이제 비특이 반응을 위해서 Blocking을 해야한다.
Phosphorylated protein을 검출하는 경우, 포스포기에 antibody가 붙기 때문에 작아서! BSA를 사용한다.
보통의 antibody는 5% skim milk를 사용하여 blocking한다. 요즘은 non protein blocking buffer가 많이 나오는데, 보통은 skim milk나 BSA로 해도 잘 된다. 시약을 판매하는 쪽에선 buffer 사용시에 blocking시간이 줄어든다고 하는데, 사실 불안해서 똑같이 돌리기 때문에 ㅋㅋ.... 크게 와닿는것이 없다.
이때 중요한 건 넉넉하게 양을 넣어 membrane이 마르거나 바닥에 막 닿지 않게, 둥둥 떠다니게 해야한다는 것이다. + swirl 형태의 shaker보다는 see-saw형태가 골고루 묻기 때문에 권장된다.
blocking 후 washing, primary antibody 붙여서 4도씨에서 overnight 한다.
6. Secondary antibody +ECL
Primary antibody 제거 후 washing, primary antibody host와 같은 host의 HRP secondary antibody를 5% skim milk에 녹여 incubation 시킨다. 이후 washing하고 ECL반응시켜 사진을 찍으면 western blot 끝!
7. Housekeeping
히히 끝인줄 알았지? 발현양을 비교하려면 Housekeeping 비교를 꼭 해야한다. Stripping buffer를 이용해 붙어있는 항체를 떼주고 5~6번 과정을 다시 시행해서 Housekeeping 단백질이 동일하게 들어가있는지 확인해준 뒤 정량해야한다.




댓글